
La malattia di Hirschsprung nei bambini non è così frequente, ma molto grave, e quindi la confusione dei genitori che sentono una tale diagnosi è abbastanza comprensibile. Il nome della malattia non è nemmeno sentito dalla maggior parte delle mamme e dei papà, e quindi ci sono più domande su questa malattia che risposte.
Cos'è?
La malattia di Hirschsprung prende il nome dal cognome del pediatra Harald Hirschsprung, che per primo descrisse la malattia dopo la morte di due dei suoi giovani pazienti per grave stitichezza cronica. Inoltre, questa malattia ha altri nomi: agangliosi del colon o megacolon congenito.
Si dovrebbe notare che la malattia è un'anomalia congenita nello sviluppo dell'intestino crasso, in cui la defecazione è estremamente difficile a causa del fatto che l'innervazione è disturbata (connessioni nervose o struttura nervosa) in una delle sezioni dell'intestino crasso, più spesso quelle inferiori. A causa del fatto che l'attività nervosa in questo reparto è disturbata, si sviluppa una diminuzione della peristalsi fino alla sua completa assenza e nelle parti dell'intestino situate sopra la zona interessata, deposizione di una grande quantità di feci.
Secondo l'OMS, la malattia di Hirschsprung nell'infanzia si verifica in un neonato su 5.000. Ci sono altri dati che suggeriscono che la malattia si manifesti con una frequenza di 1: 2000 neonati. I ragazzi sono più suscettibili alle malattie congenite: ci sono quattro piccoli rappresentanti del sesso più forte per una ragazza malata.
In 9 bambini su 10, secondo le statistiche, il megacolon congenito si trova prima dei 10 anni. In quasi un terzo dei casi, un'anomalia dello sviluppo del colon è accompagnata da altre malformazioni e ogni decimo bambino con tale anomalia ha la sindrome di Down.


Perché succede?
Il difetto è sempre congenito, ma le ragioni che lo causano sono ancora oggetto di studio da parte di medici e scienziati. Oggi, gli esperti sono inclini a credere che la formazione di un'anomalia sia influenzata da più fattori contemporaneamente, il principale dei quali è il danneggiamento di più geni contemporaneamente, che, in effetti, sono responsabili di quanto correttamente e puntualmente si formeranno le strutture neurali delle pareti dell'intestino crasso. In genetica, tutto è molto complicato e può essere difficile per i genitori capire perché, sani, hanno avuto un bambino malato. Ma proviamo a spiegare.
Fattore ereditario. Circa un bambino su cinque con un megacolon congenito ha un'influenza genetica familiare. Al concepimento, si forma un processo di mutazione nei geni RET, GDNF, EDN3, ENDRB. A 7-12 settimane di gestazione, quando il feto completa la formazione di cellule nervose nelle pareti degli organi interni, i geni interessati elencati non possono garantire la transizione delle cellule germinali-neuroblasti (da cui si formano i neuroni) nel luogo desiderato. In circa l'11-12% dei casi, i genetisti trovano aberrazioni (errori inspiegabili) nei cromosomi. E in ogni quinto bambino con la malattia di Hirschsprung, l'anomalia è strettamente associata a una sorta di sindrome ereditaria.
Anomalie a livello di sviluppo intrauterino. Si presentano nei bambini che, al momento del concepimento, avevano i geni sani sopra elencati, ma il problema e l'errore sono sorti nel processo di embriogenesi. Il motivo potrebbe essere un'infezione virale, l'influenza, subita dalla madre in una fase precoce della gravidanza, l'effetto di alte dosi di radiazioni radioattive sul corpo di una donna incinta durante questo periodo, il contatto di una donna nelle prime fasi con sostanze che possono causare mutazioni genetiche nel feto. Allo stesso tempo, la distribuzione delle cellule germinali-neuroblasti è stata interrotta.
I medici hanno seguito le statistiche e affermano con sicurezza che la probabilità della malattia di Hirschsprung nei bambini aumenta se una donna incinta soffre di varie patologie ostetriche, malattie ginecologiche, nonché gestosi, diabete mellito, ipertensione, insufficienza cardiaca o alcune malattie cardiache.


È il periodo da 7 a 12 settimane di gravidanza che è considerato critico in termini di probabilità di un fallimento nella formazione delle strutture neurali delle pareti dell'intestino crasso del feto. Fu durante questo periodo che vengono deposti i cosiddetti plessi nervosi di Meissner, che si trovano nelle persone sane nello strato dell'intestino crasso sotto la mucosa. Nello stesso periodo avviene la formazione di nodi e plessi di nervi chiamati plesso di Auenbach, situati nello strato muscolare dell'intestino.
Poiché i neuroblasti embrionali non arrivano nel posto giusto in tempo o in quantità insufficienti, le terminazioni nervose si formano con errori: i loro singoli frammenti o una rete debole e irregolare non sviluppata. Quest'area, priva del normale apporto nervoso, non può far fronte al compito principale dell'intestino crasso: la rimozione del contenuto all'esterno. Pertanto, si verifica una stitichezza costante e quindi le sezioni intestinali superiori iniziano ad espandersi.


Classificazione e tipi di patologia
I medici parleranno del tipo e del tipo di malattia in un neonato, bambino o bambino più grande in base alle caratteristiche anatomiche e cliniche dell'anomalia in un bambino particolare.
Il criterio principale è la lunghezza dell'area intestinale interessata.
Il caso più comune è la forma rettosigmoidale. Si trova nel 65-75% dei pazienti. In ogni quarto caso, si verifica la forma rettale della malattia. Solo nel 3% dei casi si riscontra una forma subtotale, nell'1,5% dei casi la lesione è di natura segmentaria. La forma totale, in cui l'intero intestino crasso è interessato e privato del supporto nervoso, si verifica nello 0,5% dei casi.
Come già accennato, le sezioni superiori si espandono a causa del costante accumulo di masse fecali e, a seconda del luogo esatto di espansione patologica, vengono aggiunte alla diagnosi specifica tali designazioni del luogo di espansione come megaretto, megasigma, megacolon subototale e totale, ecc.


La combinazione di sintomi e manifestazioni di anomalie dello sviluppo consente ai medici di identificare diversi tipi di disturbi.
- Tipico bambino - si verifica nel 90-95% dei casi. Si sviluppa rapidamente, è accompagnato da stitichezza molto frequente, quando, senza un aiuto esterno, il bambino è praticamente incapace di svuotare l'intestino, compaiono rapidamente i primi sintomi di ostruzione intestinale.
- Bambino persistente - compare per la prima volta dopo l'infanzia. Di solito ciò è dovuto al fatto che una piccola area dell'intestino è interessata e quindi il quadro clinico si sviluppa lentamente, prolungato.
- Nascosto - per la prima volta, la stitichezza dolorosa diventa un problema più vicino al periodo adolescenziale dello sviluppo del bambino. L'ostruzione intestinale è in rapido aumento.
Si distinguono anche forme compensate, subcompensate e scompensate della malattia. Se il corpo riesce a compensare parzialmente il problema, negli anni il bambino può svuotarsi, ma gradualmente diventerà incline alla stitichezza, la cui durata può variare da 3 giorni a una settimana. Con la malattia di Hirschsprung subcompensata, il bambino è in grado di movimento intestinale, ma solo con l'uso di un clistere o di un lassativo. Senza di loro, costipazione.
In uno stato di scompenso, il bambino non si fa la cacca e non sente nemmeno l'impulso a questo processo di pulizia naturale. L'ispessimento delle feci nell'intestino diventa simile a una pietra e lassativi e clisteri non aiutano.


Sintomi e segni
Dipende da quanto viene colpito l'intestino crasso quando sarà possibile determinare i primi sintomi di patologia in un bambino. Nella stragrande maggioranza dei casi, l'anomalia viene determinata dopo la nascita, ma è anche possibile un decorso latente con sintomi minori, che consentirà di diagnosticare la malattia di Hirschsprung solo a scuola, nell'adolescenza o successivamente.
Si manifesta l'innervazione dell'intestino crasso costipazione cronica. Se il bambino ha la voglia di svuotare l'intestino, ma non può farlo, non stiamo parlando di un'anomalia congenita. Con un megacolon congenito il bambino di solito non sente il bisogno di svuotare le viscere. Quasi la metà dei bambini con questa diagnosi soffre di gonfiore e dolore addominale associati all'accumulo di feci e all'espansione dell'intestino superiore.
La lunga presenza dell'anomalia forma l'apparente asimmetria dell'addome. Le vene blu delle vene sono visibili sull'addome ad occhio nudo. Il lungo decorso della malattia provoca anemia nel bambino, la sua pelle è pallida, soffre di frequenti capogiri e si stanca rapidamente. A causa dell'accumulo di feci, possono esserci sintomi di intossicazione, manifestati da mal di testa, nausea e talvolta vomito.


Diverse fasi hanno caratteristiche diverse.
- Compensato - stitichezza nei neonati, che peggiora con l'introduzione di alimenti complementari. Se i genitori monitorano la frequenza dei movimenti intestinali, somministrano clisteri in tempo, quindi le condizioni generali del bambino non vengono disturbate. Nel secondo grado, l'appetito del bambino inizia a soffrire.
- Subcompensato - la stitichezza diventa più frequente, senza un clistere diventa quasi impossibile svuotare l'intestino, ma con un trattamento adeguato, la patologia ha tutte le possibilità di andare in forma compensata. I sintomi della malattia di Hirschsprung aumentano lentamente e gradualmente.
- Scompensato - I sintomi si accumulano molto rapidamente. Al primo grado, si nota un decorso acuto: il bambino ha una ridotta scarica di gas, non c'è movimento intestinale, l'addome è gonfio come una "rana". Nel secondo grado, la malattia diventa cronica.
Un dettaglio interessante: spesso un megacolon congenito si trova nei bambini che hanno eterocromia dalla nascita - un colore diverso dell'iride degli organi visivi.
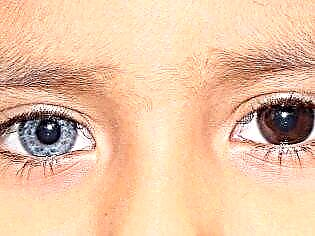

Qual è il pericolo?
L'agangliosi dell'intestino crasso è pericolosa perché i processi digestivi del bambino malato sono disturbati, i nutrienti vengono assorbiti meno del necessario e poiché tutto ciò si verifica sullo sfondo di una diminuzione dell'appetito, è possibile che il bambino diventi ipotrofico o inizi a soffrire di estremo esaurimento - cachessia.
I bambini con una tale malformazione congenita crescono lentamente, sono in ritardo rispetto ai loro coetanei in altezza e peso e spesso soffrono di disbiosi, infiammazione delle mucose intestinali.
La complicazione più pericolosa è lo sviluppo dell'ostruzione intestinale, nonché la formazione di ulcere da pressione delle pareti intestinali con calcoli fecali in violazione dell'integrità dell'intestino stesso. È pericoloso con peritonite e sepsi.


Diagnostica
Il medico può suggerire che il bambino abbia un'agangliosi intestinale sulla base dei reclami dei genitori di gonfiore frequente, difficoltà con la defecazione e costipazione. Quando un operatore sanitario tocca l'addome con le dita, vengono eseguiti due test: il test del "tumore" e il test dei "sintomi dell'argilla". Nel primo caso, il medico scopre le foche all'esame digitale dell'addome e nel secondo i segni di compressione del colon. In questo caso, il bambino viene inviato per un esame, che è progettato per confermare o negare la diagnosi. Include:
- Radiografia dell'intestino crasso riempito con mezzo di contrasto;
- endoscopia del retto e del colon sigmoideo;
- citomorfologia secondo Swenson - esame istologico di campioni di tessuto del retto e del colon;
- prove di scartamento.
Nell'analisi del sangue di un bambino con malattia di Hirschsprung, si riscontrano solitamente leucociti elevati, ESR e cambiamenti tossici nei neutrofili.
Vengono eseguiti l'esame e la diagnosi proctologi, chirurghi, specialisti in malattie infettive, pediatri.

Trattamento e prognosi
La chirurgia è generalmente raccomandata per un bambino con questa diagnosi. E i genitori dovrebbero capire che se i medici consigliano di farlo, è meglio non rifiutare, perché la malattia di Hirschsprung non può essere trattata in modo conservativo con metodi popolari. Durante l'operazione, sarà possibile ripristinare la pervietà intestinale, nonché rimuovere l'area interessata, che crea tante difficoltà nel lavoro dell'intero intestino.
Esistono due tipi di operazioni: una fase e due fasi. Per capire quale consigliare, il medico tiene conto dell'età del bambino, del grado di danno intestinale, della lunghezza dell'area innervata.
Un'operazione in una fase viene solitamente eseguita con malattia compensata e lievi danni intestinali. Basta rimuovere l'area interessata, formare un'anastomosi. Questo metodo è meno traumatico, a giudicare dalle recensioni di medici e pazienti.
Viene eseguita un'operazione in due fasi per i bambini con un disturbo nella fase di sottocompensazione e scompenso, con ampie aree colpite dell'intestino crasso. La prima fase prevede la rimozione dell'area interessata e la creazione di una colostomia temporanea. Nella seconda fase, si forma un'anastomosi.

Un'operazione di emergenza per un bambino può essere eseguita in caso di ostruzione intestinale, piaghe da decubito delle pareti, violazione dell'integrità delle pareti intestinali.
È piuttosto difficile prevedere qualcosa dopotutto, tutto dipende da quanto precocemente al bambino è stata diagnosticata un'anomalia congenita dello sviluppo, a che tipo appartiene, se tutte le raccomandazioni cliniche sono state seguite in tempo. Con un'operazione anticipata le previsioni sono favorevoli. Se il bambino non viene curato, la probabilità di morte del bambino è stimata all'80%.
Se l'operazione viene abbandonata e l'ostruzione successiva si forma, è possibile che dopo l'operazione il bambino riceva una disabilità, ma anche qui tutto è individuale e dipende dal volume dell'intervento chirurgico.

Maggiori informazioni sulla malattia nel prossimo video.


