Sulla malattia
La fenilchetonuria (PKU) è una malattia genetica associata a un disturbo metabolico degli amminoacidi, principalmente fenilalanina. L'assunzione di cibo proteico ordinario nel corpo del bambino porta all'accumulo di prodotti metabolici tossici nei tessuti. Questi composti influenzano irreversibilmente il sistema nervoso e il cervello del bambino, causando una demenza progressiva.
In alcuni paesi, la malattia prende il nome dallo scopritore ed è chiamata malattia di Völling. Un altro nome, oligofrenia fenilpiruvica, indica la patogenesi della malattia.
Le statistiche sulla diffusione delle malattie ereditarie differiscono da paese a paese. Quindi, in Russia, la malattia si trova in un bambino su 5-10 mila neonati (a seconda della regione del paese). Il paese più sicuro in termini di rischio di sviluppare fenilchetonuria è la Finlandia (1: 100.000). La Turchia è al primo posto nella comparsa di una malattia genetica, un neonato su 2600 ha un metabolismo della fenilalanina alterato.
Tra i pazienti con fenilchetonuria, le ragazze hanno 2 volte più probabilità dei ragazzi. L'ereditarietà avviene in modo autosomico recessivo, il che significa la presenza di un gene mutante nelle famiglie di entrambi i genitori. I portatori sani di un difetto genetico potrebbero non essere consapevoli delle proprie caratteristiche e potrebbero non avere manifestazioni esterne. La situazione si complica quando i proprietari della mutazione si sposano e stanno per avere figli. Il rischio di avere un bambino con fenilchetonuria in questo caso è del 25%. La metà dei bambini nati in questa famiglia rimarrà portatrice asintomatica del difetto ereditario.

Sebbene la malattia possa portare a disturbi gravi e irreversibili nello sviluppo del bambino e peggiorare significativamente la qualità della vita del bambino, è possibile evitare conseguenze spiacevoli. La fenilchetonuria è una delle poche malattie ereditarie che possono essere trattate efficacemente. Con misure tempestive prese, il bambino cresce quasi sempre sano.
Riferimento storico
Per la prima volta, la natura ereditaria della demenza è stata sospettata dal biochimico e psichiatra norvegese Ivar Asbjörn Fölling. Osservando un gruppo di bambini con ritardo mentale, il medico ha notato un quadro clinico comune e ha riscontrato un cambiamento nella composizione chimica delle urine di fratelli e sorelle malati. Aggiungendo una soluzione di cloruro ferrico al fluido biologico, il medico ha rivelato un cambiamento nel colore delle urine, l'aspetto di una tinta verde oliva. La presenza di modifiche nelle analisi in parenti stretti ha fatto sorgere il sospetto di cause ereditarie della malattia.
Questo metodo di rilevamento della fenilchetonuria non ha perso la sua rilevanza nel mondo moderno. La determinazione dell'acido fenilpiruvico nelle urine è chiamata test di Völling in onore del medico norvegese.

Come si sviluppa la malattia?
È impossibile prevedere lo sviluppo della fenilchetonuria in un neonato senza ulteriori ricerche. Il bambino non è diverso dai suoi coetanei, sembra un bambino assolutamente sano. I cambiamenti si verificano all'interno del corpo quando inizia l'alimentazione enterale, le proteine del latte materno entrano nel corpo.
La causa della patologia risiede nella mancanza di alcuni enzimi coinvolti nella conversione dell'amminoacido essenziale fenilalanina in tirosina. Pertanto, il livello di fenilalanina e dei suoi derivati nel sangue e nel liquido cerebrospinale aumenta. I prodotti metabolici hanno un effetto tossico sulle cellule cerebrali, interrompono il metabolismo dei grassi e la trasmissione degli impulsi nervosi tra i neuroni.
Allo stesso tempo, la tirosina, necessaria per il normale funzionamento, non viene sintetizzata nella quantità adeguata. Questo amminoacido è essenziale e partecipa attivamente alla formazione di ormoni, neurotrasmettitori, pigmento melanico.
Esistono diverse forme cliniche di fenilchetonuria, che sono caratterizzate da una carenza di un certo enzima nel fegato. Nel 98% si verifica la PKU di tipo I, associata a un difetto genetico nel 12 ° cromosoma. La malattia si manifesta con una mancanza di fenilalanina-4-idrossilasi e risponde bene al trattamento. I tipi II e III della malattia sono disturbi estremamente rari e sono caratterizzati da manifestazioni gravi, inefficacia della terapia dietetica nel correggere il metabolismo degli amminoacidi.
Sintomi della forma classica di fenilchetonuria
Manifestazioni comuni
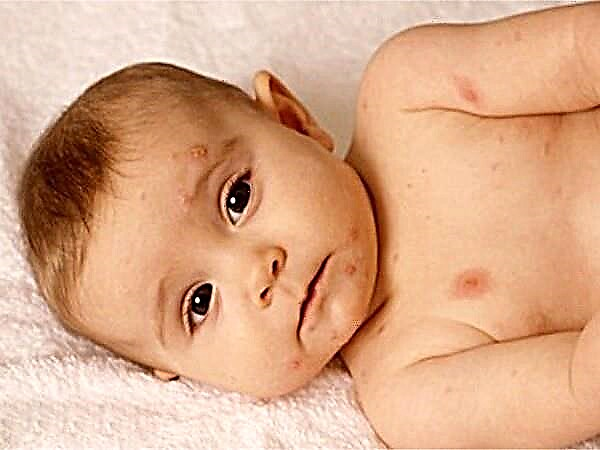
Se lo screening neonatale non è stato effettuato nell'ospedale di maternità, la malattia è rimasta non riconosciuta e le misure di trattamento necessarie non sono state organizzate, la PKU si manifesterà nel primo anno di vita del bambino. I genitori troveranno i primi segni di deviazioni dalla norma in un bambino all'età di 2-6 mesi, quando un bambino precedentemente sano diventa letargico e apatico, o viceversa, eccessivamente irritabile. Mangiare spesso finisce con il rigurgito e sulla pelle del bambino compaiono manifestazioni di dermatite allergica.
Aspetto caratteristico
Poiché la violazione del metabolismo degli amminoacidi porta ad una diminuzione della formazione di melanina, in caso di sviluppo della malattia, la pelle, i capelli e l'iride degli occhi perdono il pigmento. I bambini con una malattia genetica hanno capelli biondi, pelle sottile e pallida e occhi azzurri. Un odore caratteristico di sudore e urina di briciole può indicare una malattia ereditaria. Molti medici lo descrivono come "topo" ed è associato al rilascio di prodotti metabolici di amminoacidi con fluidi biologici.

Danni al sistema nervoso
I primi segni di cambiamenti nel sistema nervoso si manifestano con letargia, una diminuzione del tono muscolare delle briciole. Spesso si verifica distonia, compaiono contrazioni muscolari involontarie, movimenti ossessivi e convulsioni. È possibile lo sviluppo di patologie organiche del cervello: microcefalia e idrocefalo.
Quasi il 50% dei pazienti con fenilchetonuria sviluppa epilessia. A volte le convulsioni caratteristiche sono il primo segno di una malattia ereditaria. I parossismi sono difficili da trattare con gli anticonvulsivanti convenzionali e riducono significativamente la qualità della vita del bambino.
Ritardo nello sviluppo
Con il decorso della malattia, il bambino perde rapidamente interesse per il mondo che lo circonda, diventa apatico. Anche lo sviluppo fisico del bambino soffre: il bambino in ritardo padroneggia le abilità necessarie. Un ritardo nello sviluppo mentale diventa evidente quando sorgono problemi nella formazione della parola, un ritardo nello sviluppo mentale.

Gli studi hanno dimostrato che i cambiamenti grossolani nell'attività intellettuale si sviluppano dopo un anno di progressione della malattia. La briciola perde irreversibilmente fino a 50 punti QI all'anno, il che successivamente porta allo sviluppo di debolezza, imbecillità o idiozia. Pertanto, il momento in cui iniziare il trattamento per la fenilchetonuria è molto importante.
I problemi neurologici nei bambini più grandi si esprimono in iperattività, comparsa di stereotipi, movimenti ossessivi delle mani, del viso, della lingua, ondeggiamento. A volte i disturbi mentali si manifestano in stati schizofrenici.
Un grave ritardo nello sviluppo porta a una mancanza di capacità di deambulazione nel 30% dei bambini malati e alalia, sottosviluppo del linguaggio, nel 60% dei pazienti.
Varietà di patologia
Forma transitoria di iperfenilalaninemia
La malattia non procede sempre in modo classico, a volte l'aumento del livello di fenilalanina è temporaneo e la malattia stessa non si sviluppa. Un aumento a breve termine della concentrazione di aminoacidi nel sangue di un bambino può essere associato all'immaturità dei sistemi enzimatici del bambino, che è più tipica dei bambini prematuri. Questi bambini richiedono un esame più dettagliato per determinare la causa della malattia.
Varianti di fenilchetonuria dipendenti dalla pterina
Circa il 2% dei bambini a cui è stata diagnosticata fenilchetonuria in un ospedale per la maternità e a cui viene prescritta una dieta appropriata ha problemi neurologici. Questi sintomi possono indicare lo sviluppo di una rara forma di PKU dipendente dalla pterina (tipo II, III secondo la classificazione) nel bambino. Le manifestazioni cliniche delle forme atipiche di PKU sono simili al decorso della forma classica, ma i sintomi progrediscono più rapidamente e la terapia dietetica non ha l'effetto desiderato.

Un ruolo enorme nello sviluppo di forme dipendenti dalla pterina è giocato da una carenza di neurotrasmettitori, che si verifica a causa di una violazione del metabolismo degli amminoacidi. Queste sostanze sono necessarie per la trasmissione degli impulsi nervosi tra le cellule nervose, senza le quali il normale funzionamento del corpo è impossibile.
La fenilchetonuria II è considerata più maligna del tipo III. I disordini metabolici grossolani degli amminoacidi portano alla morte massiccia delle cellule nervose. Spesso i bambini con PKU progressiva di tipo II muoiono all'età di 2 o 3 anni.
Fenilchetonuria materna
I bambini con PKU non nascono sempre da genitori sani. Se le donne incinte con disturbi ereditari non seguono una dieta, c'è un'alta probabilità di avere un bambino con sindrome materna da fenilchetonuria. Le sostanze tossiche penetrano nella placenta dal corpo della madre al feto e interrompono la formazione degli organi e dei sistemi del bambino. I bambini nascono con molteplici malformazioni congenite, microcefalia, ritardo mentale e fisico.
Diagnostica
Consulenza genetica medica familiare
I genitori in attesa devono stare particolarmente attenti se nelle famiglie di qualcuno di loro ci sono stati casi di nascita di bambini con fenilchetonuria. La possibilità della nascita di un bambino con PKU non può essere esclusa, anche se la malattia si è manifestata in un lontano parente. L'insidiosità della malattia risiede nel trasporto asintomatico del gene danneggiato da parte di persone assolutamente sane.
Tutte le coppie che hanno sospetti di malattie ereditarie in famiglia dovrebbero consultare un genetista anche durante la pianificazione del bambino. Presso il Medical Genetic Center (MGC), utilizzando moderni metodi diagnostici, è possibile identificare il trasporto di un gene mutante e calcolare il rischio di avere un figlio con PKU e altre malattie genetiche.
I metodi diagnostici invasivi (biopsia corionica, amniocentesi, cordocentesi) possono aiutare a determinare la malattia prima della nascita del bambino. In questo caso, viene esaminato il materiale genetico ottenuto dal feto. Questi metodi sono traumatici e possono portare all'aborto spontaneo. Pertanto, il loro uso è giustificato solo con il trasporto comprovato di geni mutanti nei genitori e un alto rischio di PKU nel bambino.
Screening neonatale

Prima di essere dimessi dall'ospedale di maternità, i neonati vengono massicciamente esaminati per malattie ereditarie. Per questo importante studio, l'operatore sanitario preleva il sangue dal tallone del bambino e applica goccioline di fluido biologico alla parte filtrante del bianco di prova. Ogni goccia di sangue è progettata per rilevare una delle 5 malattie: fenilchetonuria, fibrosi cistica, ipotiroidismo congenito, sindrome adrenogenitale, galattosemia.
Lo screening neonatale è un esame molto importante e necessario per tutti i bambini senza eccezioni. Viene svolto in modo completamente gratuito e mira a preservare la salute della nazione. Rifiutando di manipolare, i genitori commettono un errore enorme, perché i sintomi dei disturbi ereditari non sono sempre evidenti in un neonato. Le manifestazioni cliniche chiare si verificano spesso quando è già difficile aiutare il bambino e i cambiamenti nel corpo delle briciole sono diventati irreversibili.
Questo metodo è affidabile per rilevare la fenilchetonuria, se il bambino riceve una quantità sufficiente di nutrizione enterale, latte materno. Pertanto, i neonati che si trovano nel reparto di terapia intensiva vengono analizzati in seguito. Anche i tempi del campione sono diversi nei neonati a termine e nei prematuri. Per i bambini che sono apparsi in tempo, si consiglia un set di sangue capillare il 4 ° giorno di vita. Lo studio sulle briciole nate prematuramente è posticipato a 7 giorni.
L'analisi deve essere eseguita a stomaco vuoto, il che fornirà un risultato più accurato e aumenterà il valore diagnostico del test. Il modulo con le goccioline di sangue del bambino ei dati del passaporto dei genitori viene inviato al laboratorio di consulenza genetica medica, dove viene effettuato uno studio biochimico.
I genitori devono prestare attenzione alla correttezza della compilazione del modulo, alla correttezza delle loro informazioni di contatto. Lo studio dura in media 10 giorni e la famiglia di solito è a casa a quest'ora. Se viene trovato un risultato positivo del test, i genitori dovranno contattare il centro genetico il prima possibile e le informazioni di contatto errate ritarderanno l'ulteriore esame del bambino.
Esame di un bambino nel centro di Mosca
Per confermare la malattia in un bambino e stabilirne la causa, il bambino dovrà sottoporsi a una procedura di esame in più fasi. Ripetuti test biochimici aiuteranno a confermare un aumento del livello di aminoacidi nel sangue ea prendere una decisione sulla necessità di una terapia dietetica. Viene effettuato uno studio dei livelli di fenilalanina e tirosina, l'attività degli enzimi epatici, la determinazione dei prodotti metabolici degli amminoacidi nelle urine.
Quindi viene eseguita la diagnostica genetica molecolare, che consente di identificare una mutazione nel gene RAS responsabile dello sviluppo della fenilchetonuria. Utilizzando questi metodi, è possibile determinare il trasporto asintomatico della malattia.
Trattamento PKU
Il metodo più efficace per trattare la forma classica di fenilchetonuria è considerato una terapia dietetica adeguatamente organizzata. Solo escludendo gli alimenti contenenti una grande quantità di aminoacidi dalla dieta delle briciole possiamo aspettarci risultati positivi dal trattamento.
Caratteristiche della terapia dietetica
Regole per la selezione dei prodotti alimentari
La fenilchetonuria nei bambini è una malattia difficile e solo un medico può scegliere la dieta giusta per un bambino. Nella scelta dei prodotti consentiti e nel calcolo del loro volume, il medico tiene conto di alcune caratteristiche individuali:
- quale forma di malattia è presente nel bambino;
- la gravità della malattia, il livello di fenilalanina nel sangue;
- l'età delle briciole;
- caratteristiche dello sviluppo fisico dei nervi;
- il fabbisogno fisiologico di proteine di un organismo in crescita;
- come il bambino tollera l'assunzione di una piccola quantità di fenilalanina dal cibo.
L'aminoacido fenilalanina è insostituibile, il che significa che può essere ingerito solo con alimenti proteici. Un bambino in crescita ha bisogno di un apporto sufficiente di sostanze nutritive, perché circa il 40% della fenialanina alimentare viene speso per costruire le proprie proteine. Si scopre che è anche impossibile escludere completamente i prodotti pericolosi dalla dieta delle briciole. Pertanto, nei primi giorni di terapia dietetica, si valuta in laboratorio come reagisce il corpo delle briciole ad una piccola assunzione di aminoacidi con il cibo.
L'allattamento al seno è possibile con la fenilchetonuria?
Poiché, nella maggior parte dei casi, la malattia è determinata nelle prime settimane di vita del bambino, sorge la domanda sulla sicurezza di nutrire il bambino con il latte materno. Infatti l'allattamento al seno è necessario per il normale sviluppo del bambino, la formazione del suo sistema immunitario. Ma il suo utilizzo è giustificato solo nei casi in cui la madre aderisce a regole chiare e controlla rigorosamente la quantità di cibo consumata dal bambino.
Per un'alimentazione sicura, è meglio usare latte materno appena estratto, la cui quantità consentita è calcolata dal medico in base al peso corporeo del bambino e al suo bisogno di fenilalanina.
Più piccolo è il bambino, maggiore è il suo bisogno di amminoacidi. Ad esempio, la quantità consentita di fenilalanina per un neonato è di 90-60 mg / kg di peso corporeo al giorno, mentre la quantità richiesta di amminoacido per uno scolaro è ridotta a 10-20 mg / kg di peso corporeo. La quantità di fenilalanina negli alimenti può essere calcolata considerando che 1 g di proteine contiene circa 50 mg dell'amminoacido.
Miscele specializzate
Esistono prodotti appositamente sviluppati per l'alimentazione razionale dei bambini con fenilchetonuria. Sono realizzati sulla base di idrolizzati proteici o una miscela di amminoacidi, che sono completamente o parzialmente privi di fenilalanina. La nutrizione con miscele medicinali ha lo scopo di reintegrare il corpo con le proteine necessarie, un apporto sufficiente di sostanze nutritive. Queste formule possono essere utilizzate come sostituti del latte materno o utilizzate come aggiunta alla dieta principale per i bambini più grandi.
La composizione delle miscele mediche per bambini di età diverse è diversa. Ad esempio, prodotti come "Afenilak", "Lofenalak" sono adatti a bambini di età inferiore a un anno. Ai bambini più grandi viene mostrato "Tetrafen", "Phenyl-free", "Maxamum - XP".

"Semaforo alimentare"
Ogni anno il bambino cresce e il suo bisogno di cibo aumenta. È ora di introdurre cibi complementari, provare nuovi piatti. I genitori devono ricordare i pericoli della fenilalanina per il cervello del bambino, scegliere solo alimenti approvati dal medico.
La dieta di un bambino malato è significativamente diversa da quella dei coetanei. L'alimentazione complementare inizia con l'introduzione di frutta e verdura, quindi pasta priva di proteine, vanno aggiunti alcuni tipi di cereali. Il medico curante dovrebbe comporre il primo menu del bambino e controllare il contenuto di aminoacidi nel sangue sullo sfondo dei cambiamenti nella dieta.
Tutti i prodotti sono suddivisi in gruppi in base al contenuto di fenilalanina in essi:
- lista rossa.
Si consiglia di eliminare completamente gli alimenti in questa lista in quanto ricchi di proteine. Si tratta di carne e frattaglie, pesce, uova, pane, grano saraceno, cereali d'orzo, fiocchi di Ercole;
- lista gialla.
È consentito utilizzare una piccola quantità di prodotti di questo gruppo sotto il controllo di un esame del sangue. Forse l'introduzione di semole di mais e riso, patate, latticini a basso contenuto proteico;
- lista verde.
Questi prodotti costituiscono la base della nutrizione per i bambini più grandi con fenilchetonuria: farina di mais e riso, la maggior parte delle verdure, frutta e bacche, dolci, burro.
Quando si utilizzano prodotti finiti, è necessario leggere attentamente le istruzioni sulla confezione. Mangiare banane, frutta secca e legumi può aumentare la concentrazione di fenilalanina, quindi il loro apporto dovrebbe essere limitato.

Altre terapie
L'uso di farmaci è giustificato solo per il trattamento delle forme di PKU dipendenti dalla pterina, quando la dieta non ha l'effetto desiderato. In tali situazioni vengono utilizzati farmaci L-dopa e 5-idrossitriptofano.
In altri casi, è possibile utilizzare la terapia vitaminica, i complessi minerali, come aggiunta alla dieta. Se necessario, vengono prescritti farmaci nootropici, anticonvulsivanti. Lavorare con logopedisti, psicologi, massaggi, fisioterapia hanno anche un risultato positivo nel trattamento della malattia.
Previsione e prevenzione
Il decorso della forma classica della malattia non influisce in modo significativo sull'aspettativa di vita di una persona. Ma senza un trattamento adeguato, i sintomi gravi della malattia influenzano in modo significativo l'intelligenza e la qualità della vita del paziente. In caso di terapia dietetica razionale e controllo tempestivo del livello di fenilalanina nel sangue, i bambini con PKU non differiscono dai loro coetanei e si adattano bene nella società. Le forme atipiche della malattia spesso portano alla disabilità e alla morte del bambino, poiché il trattamento raramente ha un buon risultato.
Gli esperti raccomandano di seguire una dieta per tutta la vita di una persona, ma un certo sollievo è possibile quando un bambino raggiunge i 18 anni di età. Le donne incinte con PKU devono prestare particolare attenzione per evitare la sindrome della fenilchetonuria materna in un bambino.
La prevenzione consiste nella prevenzione dei matrimoni strettamente correlati, nella consulenza medica e genetica delle famiglie a rischio per lo sviluppo di PKU e nello screening di massa dei neonati.

Conclusioni
La malattia di un bambino è un enorme stress e prova per i giovani genitori, soprattutto se la malattia è ereditaria. Ma la fenilchetonuria è una delle poche malattie genetiche per cui gli interventi terapeutici portano buoni risultati.
I genitori devono comprendere l'importanza dello screening neonatale e della terapia dietetica precoce e ben scelta. Vale la pena seguire attentamente tutte le raccomandazioni del medico curante e quindi una diagnosi così terribile come la fenilchetonuria non diventerà una frase per il bambino.